火炸药学报杂志已发表格式范文参考
1.机器学习在含能材料研究中的应用
作者:甄少聪;付小龙;韩若寒;陈启航
作者单位:西安近代化学研究所
关键词:应用化学;机器学习;火炸药;含能材料;材料设计;性能预测
摘 要:综述了近几年国内外机器学习在含能材料领域的研究进展,重点归纳了机器学习在含能材料设计和性质 预测两方面中的应用:在机器学习驱动的含能材料设计中,介绍了机器学习在分子生成方法以及辅助筛选含能分 子中的应用;在机器学习驱动的含能材料性质预测中,介绍了机器学习方法在含能材料的不同性质预测中的应用, 如爆轰性能、燃烧性能等。最后总结了机器学习在含能材料领域的深入应用面临的问题:含能材料相关的数据集 数量和数据质量问题,并对机器学习在含能材料的应用提出了展望,认为未来含能材料的发展方向是通过机器学 习结合智能机器人,建立自动化实验平台,实现“设计-预测-优化”自动化闭环优化流程。附参考文献104篇。
引 言
含能材料作为混合炸药、推进剂和发射药的主 要成分,是武器装备实现远程打击和高效毁伤的能 量来源,是武器装备发展的基础,其性能改善可能 会显著提高武器装备的射程、精度、毁伤杀伤效果 等。含能材料的更新换代可以促进武器系统的升 级换代,能够提高武器装备的动力、火力、威力,因 此新型含能材料的发现始终受到世界各国的重视。
由于含能材料的能量密度和安全性难以兼容, 且提高能量会导致材料合成更加困难,同时成本会 大大增加,这 导 致 新 型 含 能 材 料 的 开 发 和 应 用 困 难,发展受到限制。自黑火药发明以来已有很多种 含能化合物被发现,如 HMX、RDX、TNT和 CL-20等。 传统方法中研究人员通常通过试错的方式来设计 和优化含能材料的结构和性质,这种方法既耗时又昂贵,安全隐患高且人工依赖性强。为了解决上述 问题,一种基于理论计算和分子模拟,使用计算机 辅助新型含能材料研究的方法正在逐渐成为发现 含能材料的关键技术。
计算机辅助分子设计基于计算方法、预测模型 等,预先评估含能分子的爆轰性能、化学稳定性或 者与二者密切相关的其他性能。因此,它可以为实 验室研究筛选符合要求的目标分子,有效地节约经 济成本和时间成本。
近年来,人工智能在科学和工程领域取得了巨 大的突破,成为推动创新和发展的重要工具,诸如 DeepSeek、ChatGPT等人工智能大模型的出现显示 了人工智能广阔的应用前景。大数据时代也为材 料学发展创造了新的机遇[1],由数据驱动的机器学 习模型加速了“设计-预测-优化”的循环,促进了含 能材料的开发,数据驱动的机器学习有望以更低的 计算成本快速筛选材料[2]。在已有文献中,机器学 习在材料研究中发展迅速[3-9],在含能材料领域也取 得了显著的进展[10-16]。
通过对材料数据库、文献和模拟结果的分析, 机器学习模型可以发现隐藏的材料规律和新的材 料设计思路,这有助于拓展材料空间,寻找新的材 料候选者。为此,本研究概述了机器学习在含能材 料分子设计、合成及性质预测中的应用,以期为含 能材料分子设计和开发提供参考。
1 机器学习驱动的含能材料分子设计
在“设计-预测-优化”的材料开发模式下,首先 介绍的是材料的分子设计。含能材料分子设计是 指开发具有特定性能的含能分子或材料,如高能量 密度、低感度、环境友好性等性能。机器学习驱动 的分子设计是指通过生成模型或逆向设计,设计出 具有潜在高性能的新型分子结构。传统方法和机 器学习驱动的分子设计流程对比如图1所示。机器 学习方法驱动的含能材料分子设计流程包括分子 生成、数据处理、模型训练、性质预测和性能评估, 流程如图2所示。

分子生成[18]有枚举法、基于机器学习方法的生 成模型等。使用枚举法进行分子生成是指以分子 骨架为基础,枚举所有可能的取代位点使用特定基 团取代。通过系统地替换基团,可以调控化合物的 性质,例如溶解度、稳定性、反应活性等。在含能材 料领域,基团取代常用于调控能量密度、感度等关 键性能。基团取代简单高效,与机器学习结合可进 一步提升其效率和准确性,为新型含能材料开发提 供支持。在含能材料领域,现有研究中常用的取代 基有—C—NO2、—N—NO2、—O—NO2、—NH2 等,以 不同的骨架为基础,生成候选分子,再通过性质预 测模型对候选分子进行筛选。Hou [19]基于典型爆炸 物(如 TNT、CL-20、HMX、RDX等)的分子骨架,通过 枚举法生成436个分子,经过机器学习预测爆轰性 能的流程之后,使用得到的模型对这些分子进行预 测,筛选出 31 个具有优异爆炸性能的分子。Qian 等[20]以含1到6个碳原子的碳链为骨架,从枚举生 成的分子中选取1725个样本分子作为训练集,通 过性质预测的机器学习模型,综合考虑稳定性和高 爆轰性能后筛选出8个候选分子。该团队在另一项 研究中[21]基于典型的不饱和碳氢化合物骨架(如乙 烯、丙烯、丁烯等),枚举生成863个分子,基于性质 预测模型,筛选出符合爆轰性能要求的分子并进行 键解离能(BDE)计算,评估其稳定性,最终筛选出8 种高能量和高稳定性的分子。Song等[22]等使用启 发式枚举法生成3892个分子,从该分子空间中快 速选择136个符合条件的候选分子。通过大量的实 验合成,获得了8种新型含能熔铸材料,其性能实测 与预测结果吻合较好,结果如图3所示。该团队[23] 以双环碳环为基础,进行不同数量氮原子替换碳原 子,获得双杂环碳环骨架,再以硝基和氨基取代,通 过枚举法获得25112个候选分子,通过分子筛选流 程,从候选 分 子 中 快 速 筛 选 出 一 个 具 有 高 爆 轰 性 能、低感度和良好热稳定性的候选分子。

与枚举所有潜在分子的枚举法不同,基于机器 学习的分 子 生 成 方 法 可 以 学 习 分 子 生 成 规 则,如 SMILES语法,生成更合理的分子。Liu 等[24]开发了 EM-Studio平台,如图4所示,该平台中包含的分子 生成板块融合了深度生成模型和完全枚举法,如图 5所示。通过训练基于递归神经网络(RNN)的生成 模型来实现机器学习辅助的分子生成;片段对接通 过定义取代位点、枚举位点和取代基的搭配,经去 除重复结构和预优化筛选后生成分子。
Guo等[17]使用Liu开发的EM-Studio平台[24],基 于棱柱烷骨架和4种取代基[—CH3、—NO2、—NHNO2、—C(NO2)3],枚举生成了1503种分子,通过图 2流程对生成的分子进行筛选,筛选出569种爆速 不小于9000m/s的候选分子。进一步结合键解离 能(BDE)评估,最终筛选出4种兼具高能量和良好稳 定性的目标分子。Li等[25]训练了3个相关 RNN模 型(见 图 6),包 括 预 训 练 RNN、生 成 RNN 和 预 测 RNN。首先通过碎片重排和SMILES枚举的方法对初 始含能材料数据集 Dm 进行扩充。预训练RNN在扩 充的数据 集 Ds 上 进 行 训 练 以 学 习 足 够 的 结 构 知 识,通过迁移学习策略,提高生成 RNN 和预测 RNN 的模型性能。最后预测模型对生成模型生成的分子的爆轰性能进行评估筛选,筛选出45个爆速高于 RDX的分子。用高精度量子力学计算验证,发现35 个新分子的爆速比传统炸药RDX的爆速更高,并且 这些分子具有良好的热稳定性和合成可能性。

变分自编码器(VariationalAutoencoder,VAE)是 一种基于机器学习的生成模型,结合了概率图模型 和深度学习的优势,能够从数据中学习潜在表示并 生成新的数据样本。基于 VAE的分子生成方法在 含能材料领域也有应用。Balakrishnan等[26]提出局 部可优化的联合嵌入框架(LocallyOptimizableJoint EmbeddingFramework),结合变分自编码器(VAE)和 性质预测模型,工作流程如图 7所示。自动编码器 使用的是连接树自动编码器(JunctionTreeVariationalAutoencoder,JTVAE),编码器是神经网络,以分子 图为输入,将分子映射到潜在空间的分布,解码器 从潜在空间中重建分子,性质预测器从潜在向量y 中预测目标性质。通过在潜在空间中进行优化,生 成结构相似性质更优的分子。研究利用低氮分子 的丰富数据与富氮分子的少量数据进行联合训练, 用于设计富氮分子(Nitrogen-richMolecules)。该方 法结合了生成模型和性质预测模型,利用低氮分子 的大规模数据来增强富氮分子的生成模型,从而在 数据稀缺的情况下提升模型的表现。从401个富氮 分子中,优化生成了303个性质更优的分子,其中9 个分子的爆轰速度显著提升。

在筛选出符合要求的含能材料分子后,后续的 关键目标是通过逆合成策略实现目标分子的可控 制备。近年来,机器学习驱动的分子合成预测已有 研究[18,27-29]。逆合成是一种通过识别易获得且结构 简单的起始原料(前体)来设计有机合成路径的方 法,这些前体通过一步或多步反应最终转化为目标 分子。这一过程涉及化学键的断裂和官能团的转 化,使逆合成分析成为现代有机化学的重要组成部 分。与传统的逆合成策略主要依赖研究人员的经 验与知识积累不同,机器学习的引入为逆合成领域 带来了革命性的突破。尽管含能材料的逆合成研 究已取得一定进展[30-33],但机器学习驱动的逆合成 在这一领域仍处于初步探索阶段,相关应用和实践 较为有限。因此,进一步开发和研究机器学习在含 能材料逆合成中的应用,对于提升合成效率、降低 成本以及推动新型含能材料的发现具有重要意义。
在含能材料领域,合成路径往往涉及不常见或 复杂的化学反应,这些反应在现有的化学反应数据 库中数据 稀 缺,导 致 传 统 的 计 算 机 辅 助 合 成 规 划 (CASP)模型更倾向于预测常见的化学反应,而对含 能材料合成中罕见反应的预测能力有限。为了解 决这一挑战,Fortunato等[34]从美国专利文献的开放 数据集提取了253795条反应模板,通过子图匹配 (subgraphisomorphism)生成合成数据,扩展模板适用 性的训练 样 本,以 此 增 强 模 型 对 罕 见 反 应 的 适 用 性。研究对比了反应相关性神经网络(ReactionRelevanceNN)和模板适用性神经网络(TemplateApplicabilityNN)两种网络的性能。结果显示,反应相关 性神经网络对常见反应模板预测较好,对罕见反应 模板预测能力下降。相比之下,模板适用性神经网 络在罕见反应模板的识别上表现优于反应相关性 神经网络,测试集上的召回率为 0.830,精确率为 0.834。这项研究为含能材料的逆合成路径设计提 供了重要的技术突破,尤其是在识别罕见但关键的 化学反应方面。通过扩展模板适用性和改进模型 性能,该工作为含能材料的高效合成路径规划和设 计提供了更强大的工具,推动了这一领域的发展。
由于含能材料的特殊性,其合成及研究过程中 可能存在危险,未来材料开发的方向是通过人工智 能和大 数 据 分 析,促 进 新 材 料 的 发 现 和 逆 向 设 计[35,36]。智 能 制 造 结 合 自 动 化 系 统 及 智 能 机 器 人[37],实现机器人在无人干预的情况下自主合成并 筛选候选材料分子,现在已有此类平台,如合碳智能 平台(C12.ai),英国科学家研发的 AI化学家[38]。
本节总结了机器学习驱动的分子设计在含能 材料领域中的应用,基于枚举法的分子生成发展已 经十分成熟,但是其生成的分子空间中只有小部分 具有研究价值。基于深度学习模型的分子生成方 法,可以从分子数据中学习化学规律,生成符合特 定条件的 分 子,但 是 深 度 学 习 需 要 大 量 高 质 量 数 据,且对计算资源要求较高,未来有望构建高质量 数据集来进行分子生成。关于含能材料逆合成方 法,目前仍处于探索阶段,未来仍需进一步研究。
在“设计-预测-优化”的流程下,需要对生成的 分子进行性质预测和筛选,进而获得符合条件的分 子,所以接下来对机器学习在含能材料性质预测的 应用进行叙述。
2 机器学习驱动的含能材料性质预测
传统的材料研发技术是通过实验合成表征对材料进行试错和验证,而过去的计算手段受限于算 法效率,无法有效求解实际工业生产中面临的复杂 问题。近几年随着大数据和机器学习方法介入,通 过采用支持向量机、神经网络等机器学习算法训练 数据集来构建模型,以预测材料的爆轰性能、燃烧 性能等,大大推动了新型材料的发现和传统材料的 更新,预测结果甚至能够达到与仿真模型基本相同 的精度,且计算成本很低。使用机器学习预测性能 的流程如图8所示。

2.1 爆轰性能预测
含能材料的爆轰性能包括爆速、爆压、爆热、爆 温和爆容,主要用爆速、爆压、爆热来衡量含能化合 物的爆轰特性。含能化合物的爆轰性能也受密度、 生成焓、分解温度、感度等性质的影响,所以本研究 将上述性质归于此节讨论。
2.1.1 爆轰性能核心参数预测
目 前 国 外 主 要 采 用 基 于 Becker-KistiakowskyWilson(BKW)状态方程的 CHEETAN、EXPLO 等 软 件 来预估 含 能 化 合 物 的 爆 速、爆 压 和 爆 热 值。最 初 Kamlet等[39-42]使用 Ruby热化学代码生成的数据进 行训练,其模型需要以元素组成、密度和生成焓作 为输入,后续研究通过量子化学计算和经验公式来 估计密度和生成焓,并将这些参数输入到计算含能 材料爆 轰 性 能 和 热 化 学 性 能 的 软 件 中,如 Cheetah [43-51],尽管研究人员对 Kamlet模型做出改进,但 是这些改进的模型仍需要依赖密度、生成焓等参数 作为输入[52-59]。相比之下,机器学习方法可以无需 密度和生成焓即可预测爆速和爆压[60]。
2020年,Barnes [61]使用分子骨架结构(SMILES) 对定向消息传递网络(Directedmessage-passingneuralnetwork,D-MPNN)进行训练,在测试集上,模型对 爆压预 测 的 均 方 根 误 差 (RootMeanSquareError, RMSE)达到了0.626GPa,爆速的RMSE达到了0.125 km/s,和 Cheetah预测的结果相差在10%以内。研 究表明,D-MPNN 可作为快速筛选工具,替代 Cheetah运用在各种 CHNO 分子上。Casey等[62]提出一 种3D卷积神经网络(CNN),如图9所示。该模型直 接从分子的3D电子结构(包括电荷密度和静电势) 中学习,预测爆速、爆压等性质,避免了复杂特征描 述符的构建过程。这项研究首次将3D 电子结构应 用于机器学习预测分子属性,具有重要的创新意义。

2021 年,HOU 等[19]等 以 典 型 的 炸 药 如 HMX、 RDX、CL-20、TNT等为基本数据集,用非线性回归算法 中的Levenberg-Marquardt(LM)算法进行训练,预测分 子的爆速、爆压,预测结果和理论计算值进行比较, 爆速 MAE为0.3456km/s,爆压 MAE为1.4933GPa。 Huang等[63]建立了极限梯度提升回归树(XGBoost)、Adaboosting、随机森林(RandomForest,RF)、多层感知 机(MLP)和核岭回归(KernelRidegRegression,KRR)5 种模型,处理了153个含能材料的28个特征描述 符和爆轰性能,所使用 的 21648 个 数 据 通 过 高 通 量晶体级 量 子 力 学 计 算 获 得。研 究 发 现,在 5 种 模型中,XGBoost模 型 在 预 测 含 能 材 料 的 爆 速、爆 压、爆热等方面表现最佳,在测试集中爆热的 RMSE 为418.36kJ/kg,相关系数R2为0.82,爆速RMSE为 0.235km/s,R2为0.912,爆压的RMSE为1.788GPa, R2为0.91,与密度泛函理论(DFT)计算结果一致。
2023年,Guo等[17]从1503个棱柱烷衍生物分 子中选取 200 个 样 本 进 行 密 度 泛 函 理 论 (DFT)计 算,将其作为机器学习模型的训练数据,计算了分 子爆速(D)、爆压(P)和爆热(Qd)等关键性能指标, 研究使用了RF、支持向量回归(SVR)、梯度提升回归 (GradientBoostingRegression,GBR)等机器学习算法 构建模型,为分子设计提供科学依据。
也有使用密度作为特征输入来预测爆轰性能 的研究。2025年,Appleton等[64]开展了一项创新性 研究,团队训练了包含密度特征和不包含密度特征 的两 种 模 型。研 究 采 用 了 单 任 务 模 型 (Single-task Models)和多任务模型(Multi-taskModels)两种建模 策略。结果显示,多任务学习在大多数性质预测上 RMSE表现优于单任务学习,如图10所示,同时发现 使用密度作为输入特征可以提升爆轰性能预测精 度(如 Dexp),但会降低感度预测能力,如图11所示。 这项工作的创新在于首次整合实验和计算数据,通过 MT-NN实现知识迁移,缓解小数据集缺陷,同时使用 的向量选择机制可以扩展至其他材料,结合生成模型 和高通量设计,会加速含能材料的开发。本研究不仅 为含能材料的数据驱动研究提供了新思路,也为材料 科学中多任务学习的应用提供了实践范例。

2025年,Wu [65]研究收集了95种含能化合物的 实 验 爆 速 数 据,涵 盖—C—NO2、—N—NO2、—O— NO2 共3类,特征选择基于结构特征和物化性质,选 定9个易获取的特征描述符:密度、C/H/O/N 的摩 尔浓度、摩尔体积、氧平衡、活性指数、摩尔质量,采 用反向传播神经网络(BPNN)、MLP、RF和SVR4种算 法,BPNN表现最优。优化后的BPNN模型测试集误 差仅±2%,R2 为0.9852。原始数据集加入85%收 集的长氮 链 和 笼 状 结 构 数 据 组 成 新 数 据 集,修 正 BPNN为在新数据集上迁移训练的模型,新测试集为 剩余的15%长氮链和笼状结构数据,新测试集的预 测误差均小于2%,如图 12所示,显示模型广泛适 用性。该研究优点在于选取的描述符易获得,模型 扩展性很强,在新型结构(如长氮链、笼状化合物) 上也表现优秀。

不同于以上对单一种类含能化合物的爆轰性 能预测,Yang等[66]基于深度学习方法研究了混合炸 药的爆轰性能,使用人工神经网络(ANN)预测了爆 轰性能和JWL状态方程(EOS)参数。研究还构建了 一维卷积神经网络(CNN1D)模型(见图 13),输入为 炸药的爆轰参数和JWLEOS的等熵曲线,输出为炸 药的质量分数和装药密度。模型的爆速预测误差 在10% 之 内,爆 压 误 差 在 3% 以 内。预 测 的JWL EOS参数偏大,但是对应的状态方程曲线和实验值 拟合较好。该研究亮点在于逆预测模型,逆预测炸 药配方计 算 结 果 的 相 关 系 数 R2 均 在 0.99 以 上, RMSE为0.02,预测密度的 R2 为 0.9983,RMSE为 0.0107g/cm3,显示出其在工程设计中的潜在应用 价值。

从上述研究可以看出,机器学习方法在爆轰性 能研究中已展现出成熟应用,其成果显著。如表1所示,3D-CNN、D-MPNN等模型能够直接从分子结构 (如SMILES或3D 电子云数据)预测爆速和爆压性 能,大 幅 减 少 了 对 人 工 特 征 工 程 的 依 赖。 此 外, CNN1D逆模型能够根据性能指标(如爆速/爆压)反 推炸药配方,为含能材料的定制化设计提供了新的 思路。

2.1.2 密度预测
由于K-J方程中爆轰速度与装药密度正相关且 爆轰压力与装药密度的平方成正比,因此装药密度 被认为是影响含能材料爆轰性能的关键参数。在 新材料的研发中,常以材料的晶体密度近似装药密 度,来设计筛选出潜在的高爆轰性能化合物。
定量结构-性质相关(QuantitativeStructure-PropertyRelationship,QSPR)研究是一种寻求分子结构与 化合物性质潜在关系的方法。在完成 QSPR模型的 建立后,计算时仅需要输入分子的结构信息,无需 其他实验参数即可预测相关新的,甚至未合成的化 合物的性质[67]。2021年,Yang等[68]建立了基于分 子拓扑结构的图神经网络模型,用于预测含能化合 物密度。研究采用了支持向量机(SVM)、随机森林 (RF)和图神经网络(GNN)3 种模型。以 Cambridge 结构数据库中超过2000个已报道的硝基化合物为 训练和 测 试 数 据。结 果 表 明 (见 图 14 和 图 15), SVM 和 RF模 型 仅 使 用 指 纹 位 向 量 (fingerprintbit vectors)时误差较大。相比之下,GNN模型结合分子 拓扑结构能够更准确地预测密度,其准确性略高于 耗时 较 长 的 DFT-QSPR 方 法。 在 独 立 测 试 集 上, SVM、RF、DFT-QSPR 和 GNN 模 型 的 预 测 误 差 小 于 5%的化合物比例分别为48%、63%、85%和88%。 因此,基于 GNN和分子拓扑结构的模型具有更高的 准确性和较低的计算成本,更适用于高通量筛选高 能化合物。
2021,Chen等[69]开发了两种新的特征化方法体 积 占 据 空 间 矩 阵 (volumeoccupationspatialmatrix, VOM)和热贡献空间矩阵(heatcontributionspatialmatrix,HCM),分别来预测晶体密度和固相生成焓。以最 优模型KRR为例,使用 VOM 特征比使用BOB特征的 平均绝对误差从0.048g/cm3减少到0.035g/cm3,通 过交叉验证,该方法适用于除立方体外的大多数含 能材料,显著提高了预测精度。Nguyen等[70]结合机 器学习模型评估了固定特征和学习特征两种不同 的特征(见 图 16),固 定 特 征 使 用 RDKit描 述 符 和 E3FP指纹,学习特征是通过图神经网络学习分子表 示,仅使用 原 子 和 键 的 信 息,自 动 提 取 相 关 特 征。 研究结果显示,基于学习特征的 MPNN模型表现最 佳,在训练 集 之 外 的 其 他 数 据 集 测 试 也 能 表 现 优异,展现了强大的泛化能力。


将 QSPR方法和机器学习结合可以用来预测材 料性质。Fathollahi等[71]利用 QSPR方法研究了高能 共晶(ECs)的密度(ρ)。开发了利用相同的描述符建 立人工神经网络(ANN)模型和多元线性回归(MLR)模 型。ANN和 MLR模型的相关系数(R2)分别为0.9716 和0.9309,ANN在完整数据集的平均绝对相对偏差 为2.48%,表 明 ANN 更 高 的 准 确 性 和 可 靠 性。Li 等[72]结合了 MonteCarlo(MC)和变量重要性测量 (VIM)改进了随机森林 (RF)方法,使用定量构效 关系 (QSPR)选择特征描述符,利用高斯软件对162 个含能化合物进行结构优化,并根据优化后的分子 结构,利用 CODESSA软件计算分子描述符数据,构 建的 MCVIMRF_Med模型如图17所示。结果表明, MCVIMRF_Med集成模型对含能物质密度具有较好 的预 测 效 果,预 测 结 果 R2 为 0.9768,RMSE 为 0.0578g/cm3,该模型是一种有效的含能材料密度 预测方法。Zohari等[73]采用 QSPR方法研究了含能 唑类化合物密度与其分子结构的关系。通过多元 线性回归(MLR)模型将唑类含能化合物的密度与化 合物最佳元素组成、化合物的不饱和度(DoU)、结构 式中的亚硝基等参数相关联,得到关联式的均方根 偏差(RMSD)为0.038g/cm3。

Readcross(RA)是一个根据近源化合物和查询 化合物之间的相似性来预测分子的性质的平台,将 RA和 QSPR结合,可以得到q-RASPR(定量跨结构-性 质关系)模型,与传统的 QSPR技术相比,q-RASPR方 法具有较好的泛化性能,与机器学习方法结合,可 以提高模型的预测能力[74]。
2023年,Jin [75]提出了力场启发式 Transformer网 络(FFiTrNet),通过融合分子力场能量项与Transformer 架构,显著提升了笼状分子密度预测精度。模型在 CSD测 试 集 上 表 现 最 优 (MAE=0.0313g/cm3),在 Huang&Massa数据集[76]外部测试集上仍保持最佳 泛化能力(MAE=0.0489g/cm3)。误差增加主要源 于训练数据(CSD)缺乏高密度和含 Cl样本。
近年来,含能材料密度的机器学习预测研究取 得了显著进展。Casey [62],Barnes [61],HOU [19]等也对 密度预 测 有 所 研 究,研 究 结 果 总 结 见 表 2。传 统 QSPR方法通过与机器学习结合(如 ANN、MCVIMRF _Med等模型),实现了对高能共晶、唑类化合物等密 度的精准预测。更创新的方法如基于分子拓扑结 构的 GNN(误差小于5%)、VOM/HCM 空间矩阵特 征(MAE降低至0.035g/cm3),以及 MPNN的自动特 征学习,进一步提升了预测精度和泛化能力,其中 Casey基于 分 子 3D 结 构 的 3DCNN 模 型 误 差 最 低 (RMSE为0.014g/cm3)。然而,现有研究仍存在局 限性,比如特征工程(如 VOM、分子描述符筛选)的 计算成本较高,制约了高通量筛选效率。未来研究 可探索多模态数据融合(如结合晶体学数据库与量 子化学计算)、轻量化深度学习架构,最终服务于含 能材料的设计。

2.1.3 生成焓预测
生成焓是指在标准大气压下、298.15K时的生 成焓,其定义为最稳定单质按照化学计量比形成某 一物质的反应焓变。生成焓是预测含能材料爆轰 性能的重要参数。
2022年,Mathieu等[77]使用 ANI-1X模型预测分 子几何构型和振动频率,使用 ANI-1CCX 模型预测 电子能量,采用原子当量法(AtomEquivalent,AE)计 算生成焓。对烃类和普通有机分子,ANI模型优于 DFT方法,MAE在17~30kJ/mol之间,与已发表文 献吻合,但是对含能材料(如硝基、叠氮基化合物) 预测误差较大(RMSE=32.5kJ/mol),这是因为训练 数据缺乏 不 稳 定 基 团 (如 硝 基、过 氧 化 物、叠 氮 基 等),导致模型无法准确外推这些结构的生成焓,ANI 模型和其他模型在含能材料数据集上的误差对比 如图18所示。

2024年,Zhang等[78]对比了 RF、MLP、图卷积网 络(GCN)与消息传递神经网络(MPNN)模型,其中 MPNN 模 型 表 现 最 好 (RMSE=35.25kJ/mol,R2 = 0.992),MPNN 无需人工设计描述符即可达到高精 度,为高通量筛选含能分子提供新范式。
升华焓是含能化合物的一个重要性质,用于推 导固相生成焓并进一步预测爆轰性能。Liu等[79]通 过皮尔逊相关性分析筛选出与升华焓相关性强的4 个基于分子拓扑结构的描述符(A、TPSA、nRNO2、S), 构建了 QSPR方程,并根据方程构建粒子群优化算 法(ParticleSwarm Optimization ,PSO),并训练机器 学习模型。结果表明,XGBoost模型预测精度最高, 平均绝对误差(MAE)为11.3kJ/mol,PSO 模型虽然 精度略低于 XGBoost,但其完全可解释的函数形式 使其更具可移植性和推荐价值。
此外 Guo [17],Casey [62],Barnes [61]等也对生成焓 进行了预测,结果见表3。生成焓的精确预测对含 能材料性能评估有关键意义,然而,现有方法仍存 在显著局限性:Mathieu等的研究虽采用机器学习方 法,但与 DFT计算结果相比仍存在较大偏差(见图 18)。另一方面,Guo的预测模型虽在现有研究中表 现出最优精度,但其训练数据仅涵盖棱柱烷类衍生 物,对其他含能材料体系(如硝基化合物、金属有机 框架等)的适应性尚未验证。综上所述,生成焓的 预测仍需进一步研究,需要建立含能基团专项数据 库,进一步开发机器学习模型,促进生成焓的预测 研究。

对含能材料分子进行筛选,除了考虑高能量, 还要兼顾稳定性,衡量含能材料稳定性的指标有感 度、热分解温度、键解离能等。
2.1.4 热分解温度预测
热分解温度是含能材料在加热过程中开始分 解的温度值,这一参数对含能材料在储存、运输和 应用中的安全风险评估至关重要。
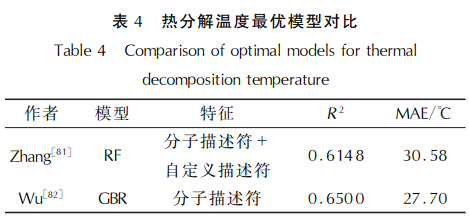
Fathollahi等[80]使用人工神经网络 (ANN,采 用 Levenberg-Marquardt算法)和多元线性回归(MLR)方 法,基于 Dragon软件生成的分子描述符,预测了30 种高能共晶体的分解温度。研究结果显示,ANN模 型在测试集上的预测 R2 达到了0.9802,平均绝对 偏差1.69%,显著优于 MLR 模 型 (R2 =0.7438)。 这一结果表明,ANN模型在处理非线性关系时具有 更高的预测精度,其优异的性能为高能材料的安全 性评估和性能优化提供了可靠的理论支持。Zhang 等[81]对比了KRR、LASSO、RF、XGBoost等7个模型预 测热分解温度的性能。结果表明,RF模型预测效果 最佳,R2 为0.6148,MAE为30.58℃。Wu [82]比较 了特征选择对训练结果的影响,表明分子描述符表 现优 于 ECFPs,同 时 对 比 了 弹 性 网 络 模 型、SVM、 KNN、MLP和 GBR模型,GBR模型表现最佳,测试集 的R2 值 为 0.65,MAE为 27.7℃,显 著 优 于 其 他 模型。
热分解研究是含能材料安全性评估的重要环 节,但目前机器学习方法在该领域的应用仍存在一 定局限性,如表 4所示,目前,机器学习在该领域的 应用仍面临诸多挑战。现有研究虽然尝试建立热 分解温度预测模型,但普遍呈现预测精度不足的问 题。深入分析发现,现有研究的特征选择存在明显 局限性:Zhang采用分子描述符结合自定义参数(氧 平衡、元素含量、硝基数量等)的建模方法,Wu则主 要依赖分子描述符。这些方法虽然考虑了分子内 特性,但忽视了诸如分子间相互作用(氢键、范德华 力等)、晶格能等关键因素,这可能是导致预测偏差 的重要原因。
2.1.5 感度预测
在含能材料开发中,能量与感度之间的平衡是 一个核心挑战。能量越高,材料在冲击、摩擦、火焰 或静电刺激下发生燃烧或爆炸的可能性越大,即感 度越高、安全性越低。因此,如何设计出兼具高能 量与低感度的含能材料,成为该领域的关键科学问 题。感度作为衡量含能材料安全性的重要指标,其 预测在含能分子研究中具有至关重要的作用。通 过结合爆轰性能预测模型与感度预测模型,可以有 效筛选出 具 有 高 能 量 且 低 感 度 的 理 想 含 能 材 料。 目前,已有 研 究 采 用 传 统 方 法 构 建 了 感 度 预 测 模 型[83-85],然而,利用机器学习方法预测含能材料感 度的研究仍处于起步阶段,尚存在较大的探索空间。
早至2007年,Keshavarz等[86]就使用人工神经 网络(ANN)来预测含能分子的撞击感度(H50),该模 型在训练集和测试集上的均方根误差(RMSE)分别 为41cm和56cm,表明其能够有效捕捉高能分子结 构与撞击感度之间的非线性关系。但是模型的误 差较大,只能为实际应用提供一定的参考价值。
2021 年,Deng 等[87] 使 用 了 人 工 神 经 网 络 (ANN)和确定独立筛选和稀疏化算子(SISSO)方法, 构建了针对240种硝基芳香化合物的预测模型。结 果表明SISSO 的平均RMSE值为(0.94±0.19),ANN 的平均 RMSE 值 为 (0.86±0.18),t-test结 果 为 -1.83,表明差异不显著。也就是说,SISSO 模型在 映射冲击敏感性和体积模量之间的关系方面与人 工神经网络表现出相当的性能。
2023年,Duarte等[88]通过量子化学计算与机器 学习相结合,定量分析分子电子结构对撞击感度的 贡献,采用密度泛函理论(DFT/B3LYP)计算分子电 荷密度,通过分布式多极分析(DMA)分解为特征, 训练多种机器学习模型,测试集上 GradientBoosting 的RMSE 最 低,为 27.5cm。 但 是 样 本 量 较 小 (53 种),且只有硝基芳香族化合物,未来需扩展至其他 炸药家族(如硝胺类)。
2024年,Wu等[89]对比4种机器学习模型——— 反向传播神经网络(BPNN)、多层感知器(MLP)、随机 森林(RF)和支持向量回归(SVR),预测了含能化合物的撞击感度和静电火花感度,结果如图 19所示, 其中BPNN模型表现最佳。

2025年,Liu等[90]基于密度泛函理论计算和文 献数据,采用4种机器学习算法(AdaBoost、SVR、RF、 KRR)对撞击感度进行预测。RF模型在预测撞击感 度[ln(H50)]方面表现最佳,测试集对ln(H50)预测的 RMSE为0.55,R2 为0.74,如图 20所示。其研究 发现撞击感度受爆热、氧平衡、分解产物、HOMO 能 级影响较大。

对于大多数含能分子而言,其感度与分子中特 定化学键的 稳 定 性 密 切 相 关,尤 其 是 碳 硝 基 (C— NO2)、氮硝基(N—NO2)和氧硝基(O—NO2)键的离 解能。这些化学键的强度直接影响含能分子在外 部刺激(如冲击、摩擦、火焰或静电)下的分解行为, 键解离能越低,化学键越容易断裂,含能材料的感 度越高,稳定性越低;反之,键解离能越高,材料的 稳定性越强,感度越低。因此,键解离能可以作为 评价含能材料稳定性的替代指标,为感度预测提供 理论依据。
2024年,Gou等[91]收集了778种真实的 CHON 含能材料,并通过量子力学方法(B3LYP/6-31G** ) 计算了其BDE值,构建了一个具有多样性和代表性 的数据集。数据集涵盖了多种含能骨架和取代基, 为模型的泛化能力奠定了基础。在特征选择方面, 研究采用了混合特征策略,见图 21(a),包括全局特 征(SEC),结合了SumOverBonds(SOB)、电拓扑状态 指纹(E-state)和自定义描述符集(CDS),能够全面描 述 分 子 的 整 体 性 质;局 部 特 征:化 学 键 描 述 符 (CBD),重点捕捉分子中关键化学键的局 部 信 息。 实验结果表明,混合特征(SEC+CBD)的表现显著优 于单一特征表示,这验证了全局与局部特征结合的 有效性。此外,研究提出了一种创新的数据扩充策 略———PairwiseDifferenceRegression(PADRE),见 图 21(b)。该方法通过增加数据的大小和多样性,显著 提升了模型的鲁棒性和预测精度。结合混合特征 和PADRE数据增强后,XGBoost模型的性能达到了 R2=0.98和 MAE=8.8kJ/mol,展现了极高的预测 能力。

对于模型的可解释性问题,可使用特征重要性 排序、SHAP分析、模型可视化工具、领域知识和模型结合等。SHAP(SHapleyAdditiveexPlanations)是一种 基于博弈论的模型解释方法,用于解释机器学习模 型的预测结果。它结合了Shapley值理论,将每个特 征对模型输出的贡献公平分配,从而提供透明且可 解释的特征重要性分析。特征重要性排序常使用 随机森林算法,随机森林在拟合数据后,评估每个 特征在随机森林每棵树上的贡献,取平均值作为贡 献值,比较不同特征的贡献值进行重要性排序,重要 性排名靠前的特征对预测结果影响更大。将领域知 识和模型结合,Liu [79]依据特征描述符构建 QSPR方 程,并根据方程构建粒子群优化算法(ParticleSwarm Optimization,PSO),增加了模型的可解释性。
模型预测性能的好坏与特征选择密切相关,如 表5所示,Gou [91]的研究表明,选择合适的特征组合 能够显著提升模型的预测精度,田小兰[92]也研究了 特征选择对预测结果的影响。尽管机器学习在含 能材料性质研究中取得了显著进展,但仍面临诸多 挑战:首先,含能材料的实验数据规模相对有限,这 直接制约了模型的训练、优化及泛化能力的提升。 其次,当前 研 究 中 主 要 关 注 的 含 能 材 料 多 限 于 含 碳、氢、氧、氮元素的化合物,然而,含氟、氯等元素 的含能化合物以及离子型含能材料等同样具有重 要研究价 值。仅 聚 焦 于 C、H、O、N 体 系 的 含 能 材 料,难以全 面 揭 示 复 杂 含 能 材 料 的 多 样 性 及 其 特 性。这些问题不仅限制了机器学习在含能材料研 究中的潜力,也阻碍了该领域的进一步发展。

2.2 燃烧性能预测
燃烧性能有别于爆轰性能,常说的燃烧性能主 要是指固体推进剂的燃烧性能,是固体推进剂的重 要性能之 一,其 主 要 包 括 燃 速、燃 速 压 力 指 数 等。 其中核心参数是推进剂的燃速,无论燃烧时压力、 初温、气流速度等如何变化,最终都反映在燃速的 变化上,所以必须研究燃速及燃速的变化规律,为 装药设计者推进剂配方设计提供参考。
传统研究多采用配方组分的种类、含量和粒度 等直接参数作为特征输入。王丽莹等[93]比较了RF、 支持向量回归(SVR)等机器学习方法的性能。结果 表明,以多项式内积(Poly)为核函数的支持向量回 归(SupportVectorRegression,SVR)模型的预测效果 最优,其模型的留一法交叉验证结果 R2为0.9927, RMSE为0.5553mm/s。
郭延芝等[94]基于王丽莹的预测模型,对含RDX 的改性双基推进剂进行了燃速性能的智能化配方 优化与设计。在给定压强下在参数空间进行搜索 迭代,筛选出燃速最高的配方。研究发现差分进化 算法优化改性双基推进剂燃速性能方面表现最佳。 该团队[95]在另一项研究中提出基于多任务学习的 机器学习策略,综合考虑推进剂组分、含量、压强和 粒度对目标性能的影响,比较了10种机器学习算法 的建模效果。其中,XGBoost模型预测性能最优,预 测燃速的R2为0.9985,RMSE为0.2571mm/s,在对 6个外部样本的测试中,模型对燃速的预测误差均 在5%以内。Pang等[96]通过人工神经网络(ANN)预 测了固体推进剂的燃速,得到测试集中71%的RMSE 为5.1×10-4mm/s,研究通过输入燃速反推压力或 组分比例,对配方进行优化。但是逆向任务的多解性存在误差,需要进一步优化。孙娜等[97]对多个模 型进行融合,预测了丁羟推进剂的燃速,融合模型 在测试集上的 RMSE为0.106mm/s,在样本集外的 10组数据上的RMSE也达到了0.321mm/s。陈少臣 等[98]建立了对改性双基推进剂的机器学习模型,表 现最好的SVR模型RMSE为0.828mm/s。
不同于通过模型直接预测燃速,Klinger等[99]使 用了直接预测燃速和预测a 和n,再以经验公式预 测燃速的两个方法,以收集了超过601个公开可用 的固体推进剂配方的材料组成和燃烧速率参数,构 建了一个完整的数据集,分别以公式u=aPn 中的 u、a、n 作为预测目标,建立3个随机森林机器学习 模型,利用这个大量的公开数据来预测固体推进剂 的燃烧速率参数,预测结果与已有结果相比均十分 接近,如图22所示。

相较于传统的特征表示方法,张小平等[100]利 用遗传(GA-BP)神经网络建立了 NEPE 固体推进剂 燃烧模型,通过全新的配方表征方法预测高压燃烧 性能,该研究将影响燃烧的元素含量(C、H、O、N、Cl、 Al)、反应物总生成焓固体组分的分形维数和固体组 分粒度参数作为输入特征。测试结果表明,预测燃 速的相对偏差小于5%的占91.7%,相对偏差大于 10%的仅占1.04%,说明系统准确率高,能满足高 能推进剂高压燃速计算需要。
当前固体推进剂燃速预测的研究已从传统经 验模型转向数据驱动的智能算法,通过整合配方成 分、压力、粒度等多维度特征,显著提升了预测精度 与泛化能力。主流方法中,XGBoost模型表现优异, R2 可达0.99以上,RMSE低至0.26mm/s。此外优 化算法(如差分进化算法)的引入使得模型不仅能 预测燃速,还可以反向设计高燃速配方,为推进剂 的智能化设计提供可能。然而,部分研究结果的解 读需谨慎。Pang等[96]报告了71%测试样本的RMSE 达到了为5.1×10-4,但在完整数据集上的表现未 知。此外,数据量是制约模型精度进一步提高的因 素之一,研究所用的数据集规模都不大,见表6。与 通过实验获得的数据不同,Klinger等[99]从文献中收 集数据,推进剂配方信息会有缺失,这影响了数据 集的质量,进而会影响模型预测精度。另外小样本 数据集数据增强后可能引入偏差,而Pang的研究中 的多解性问题(如逆向预测压力/组分)仍需解决。 未来研究需建立大数量、高质量燃速数据库,还需 要开发合适的模型,比如开发多任务学习模型,同 时预测燃速、压力指数等燃烧性能,促进数据驱动 的推进剂配方设计,为推进剂数字化设计提供更可 靠的工具。

除了上述性质之外,机器学习在其他方面也有 着应用。如集成光流控和机器学习的典型含能化 合物微观结晶热力学研究[101]、用机器学习分子动 力学模 拟 研 究 高 能 轰 击 后 晶 体 缺 陷 的 分 类 和 量 化[102]、金 属 吸 附 能 预 测[103]、材 料 力 学 性 能 预 测[104]等。
由上述研究可知,机器学习方法在含能材料性 质预测中已经有了成熟的应用,能够解决大部分性 能预测问题,为含能材料结构设计筛选和优化奠定 基础。
3 结论与展望
本研究表明,机器学习在含能材料领域中的应 用已取得显著进展。基于“设计-预测-优化”闭环循 环的含能材料开发模式已逐步投入实践,机器学习 在整个流程中发挥了关键作用。然而,机器学习在 含能材料领域的应用仍面临诸多挑战,主要包括以 下方面:
(1)数据数量不足:含能材料研究在材料科学 中占比较小,导致相关数据的积累相对有限,且由 于保密性 要 求,许 多 含 能 材 料 的 实 验 数 据 无 法 公 开,因此导致了关于含能材料的数据样本量少且种 类分布不均等问题。这种数据稀缺性限制了机器 学习模型的训练效果与性能。缺乏多样性和代表 性的数据可能导致模型出现过拟合或欠拟合现象, 难以准确捕捉含能材料中的复杂关系和特征,从而 影响模型的预测能力与泛化性能。
(2)数据质量问题:现有文献中的数据往往存 在不完整性,例如推进剂配方中缺少组分或含量信 息,反应路径中缺失中间体或关键步骤等。此外, 实验数据易受环境影响,相同实验在不同条件下可 能产生差异较大的结果。这些问题导致数据质量 参差不齐,异常数据的存在会显著降低机器学习模 型的预测准确性。
为了深化机器学习能在含能材料领域的应用, 未来的研究方向建议从以下几方面开展:
(1)针对数据量不足问题,开发数据增强技术, 充分挖掘文献数据中含能材料结构与性能之间的 相互关系,结合迁移学习等手段,突破数据数量限 制;通过高通量量子化学计算和分子动力学模拟, 生成涵盖不同性质含能材料的标准化海量数据集, 提升模型预测的可靠性。
(2)建立机器学习-智能合成机器人协同平台, 集成文献 挖 掘 分 析、分 子 结 构 设 计 和 合 成 路 线 设 计、自动化合成验证3个模块,实现虚实结合的含能 材料智能化研发,提高新型含能材料的研发效率。
当前,各种机器学习模型的成功应用证明了其 在含能材料领域的光明前景,基于机器学习方法的 自动化材料开发方法代表了未来含能材料研究的 重要发展方向。随着数据积累与技术进步的不断 推进,人工智能有望在这一领域发挥更加重要的作 用,推动含能材料研究的突破性进展。
